赵奶奶曾不小心摔倒过一次,导致手臂骨折,住院治疗一段时间后伤势逐渐好转。但陈爷爷却发现,自那以后自己老伴的记忆出现了问题,总是忘东忘西,有时候看到熟人甚至也有点反应不过来。一开始,他以为是妻子摔伤受到了惊吓,直到她连门牌号都记错,陈爷爷才觉得问题没那么简单。
去医院检查后,赵奶奶确诊了阿尔茨海默病,也就是人们俗称的老年痴呆。陈爷爷有很长一段时间没办法接受这个事实,他想不通原因。后来,女儿和他解释以后他才大概明白,妻子的病是脑子里出现了“病变”。每个患上阿尔兹海默病的人,脑海里都有一块“橡皮擦”,他们仿佛成了遥远的旁观者,在循环往复的混乱世界里,一次又一次地迷失着自我。
阿尔茨海默病(AD),俗称“老年痴呆症”,是最常见的神经退行性疾病。
AD的特征是大脑皮质及特定皮质下区域的神经元和突触丧失,神经元丧失过多甚至会导致区域性脑萎缩[1]。其病理特征包括老年斑(senile plaques, SP)和神经纤维缠结(neurofibrillary tangles, NFTs),二者分别由大量沉淀的β淀粉样蛋白(Aβ)和过度磷酸化的Tau蛋白组成。
AD可以分为早发性AD(early-onset AD,LOAD)和晚发性AD(late-onset AD,LOAD)。两者之间的区别在于,LOAD患者一般会携带一些遗传性致病基因,三个最常见的AD突变是APP基因突变、PSEN1基因突变和PSEN2基因突变[2]。目前几乎所有用于研究AD的动物模型都是建立在该疾病的遗传变异的基础之上,将已发现的AD突变基因引入小鼠的基因组中成为这些研究模型的建立基础[3]。

图1 AD小鼠动物模型
尽管目前已经有很多的转基因小鼠动物模型被用来研究AD,但这些小鼠模型通常是由人工诱导的,无法完全表征人类AD的病理表现。比如在常见的5XFAD转基因小鼠模型中,并不能观察到tau过度磷酸化和NFTs的形成[4];而双转基因APP/Tau小鼠和三转基因APP/Tau/PSEN转基因小鼠模型[5]尽管可以较好地重现AD的典型病理特征,但却难以解释这些病理特征是如何相互关联的[6, 7]。也因此,关于Aβ病理是否可以诱导tau缠结以及神经元如何在阿尔茨海默病中死亡的基本问题一直没有得到解答。
2023年9月14日,比利时VIB-KU鲁汶脑与疾病研究中心的 Bart De Strooper 团队在国际顶尖学术期刊 Science 上发表了题为 MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’ s disease 的文章。
他们选择了一种新的AD小鼠模型——即将人类神经元异种移植到小鼠体内,回答了Aβ病理是否可以诱导tau缠结以及神经元如何在AD中死亡的问题,揭示了人类神经元对阿尔茨海默病的独特易感性,也为AD的防治带来了全新的潜在靶点和方法。
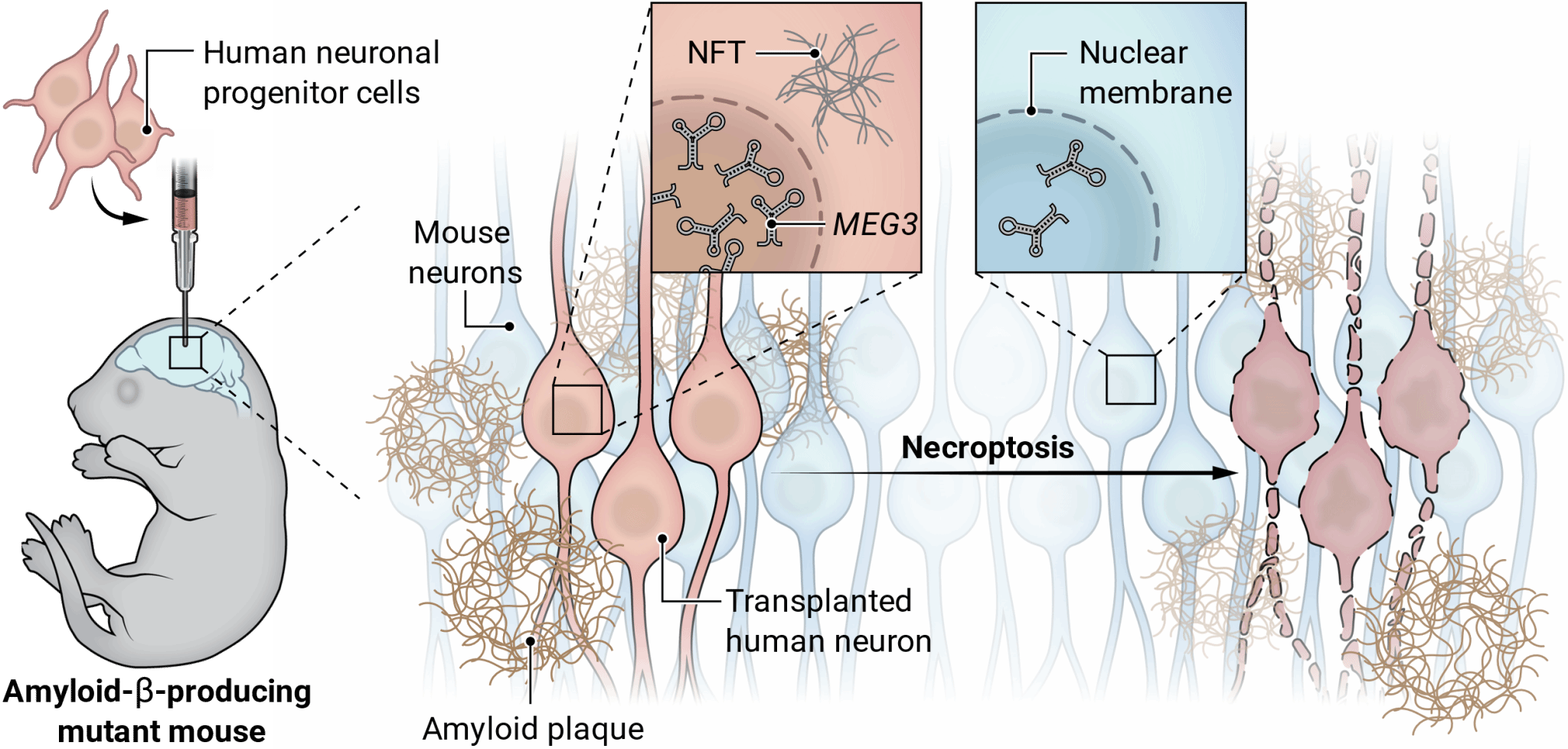
图2 移植到淀粉样变性小鼠模型中的人 源 神经 元 最终发展为tau病理并 发生 坏死
1、淀粉样斑块沉积足以在人源神经元中诱导病理性tau
研究团队首先基于先前的Nod-SCID小鼠的异种移植模型作出了改进,他们使用Rag2-/- (Rag2tm1.1Cgn) 免疫抑制遗传背景和单个AppNL-G-F (Apptm3.1Tcs/Apptm3.1Tcs) 敲入基因来驱动Aβ病理,构建了Rag2-/-/AppNL-G-F淀粉样蛋白小鼠模型以接受人类干细胞来源的神经祖细胞(NPC)移植。
移植2个月后,异种移植神经元已经能够表现出成熟神经元特征和皮质标记物。移植18个月后,小鼠出现了完全的淀粉样斑块病理;与移植小鼠神经元的对照组相比,移植人类神经元的小鼠表现出了严重的AD病理,包括神经纤维缠结(NFTs)、颗粒空泡性神经变性(GVD)和神经元丢失。这表明存在未知的人类特异性特征定义了人类神经元对Aβ病理的敏感性。
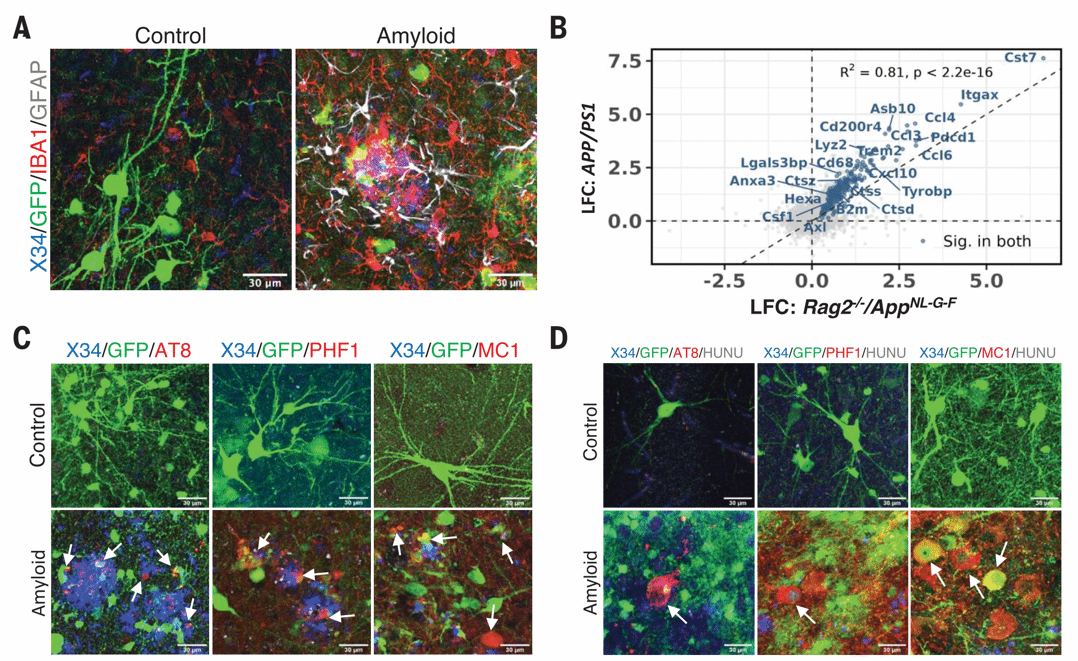
图3 淀粉样斑块沉积足以在人源神经元中诱导病理性tau
2、移植人源神经元发生坏死性凋亡,测序发现MEG3显著上调
定量PCR结果表明淀粉样蛋白小鼠中约50%的移植人源神经元丢失,那么这些暴露于淀粉样蛋白病理的人源神经元是如何死亡的呢?
为了回答这个问题,研究者分离出了移植后2、6和18个月时小鼠的异种移植人源神经元,进行RNA测序。研究者并未观察到与细胞凋亡或铁死亡有关的基因表达变化,但结果显示程序性坏死执行蛋白MLKL表达上调,qPCR检测AD患者颞回脑样本也验证了MLKL和RIPK3的显著上调,证实了程序性坏死的存在。
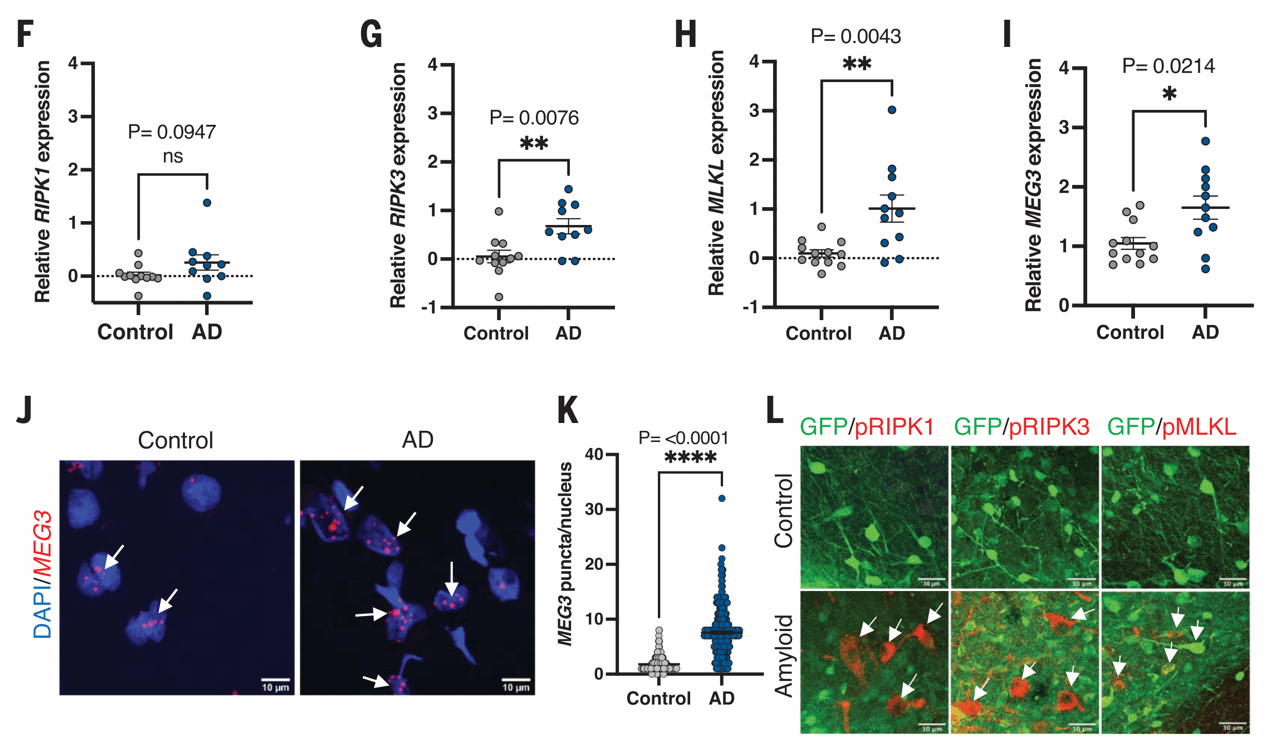
图4 移植人类神经元发生程序性坏死
那是什么在驱动神经元程序性坏死呢?研究者在暴露于淀粉样蛋白的异种移植人类神经元中发现了数十个表达改变的长链非编码RNA,其中上调幅度最显著(超过10倍)的MEG3引起了研究者的关注。MEG3通过p53参与细胞死亡途径,虽然此前从未有研究认为MEG3参与AD,但研究者在单细胞核转录组数据库中发现,AD患者大脑中存在MEG3上调的现象。
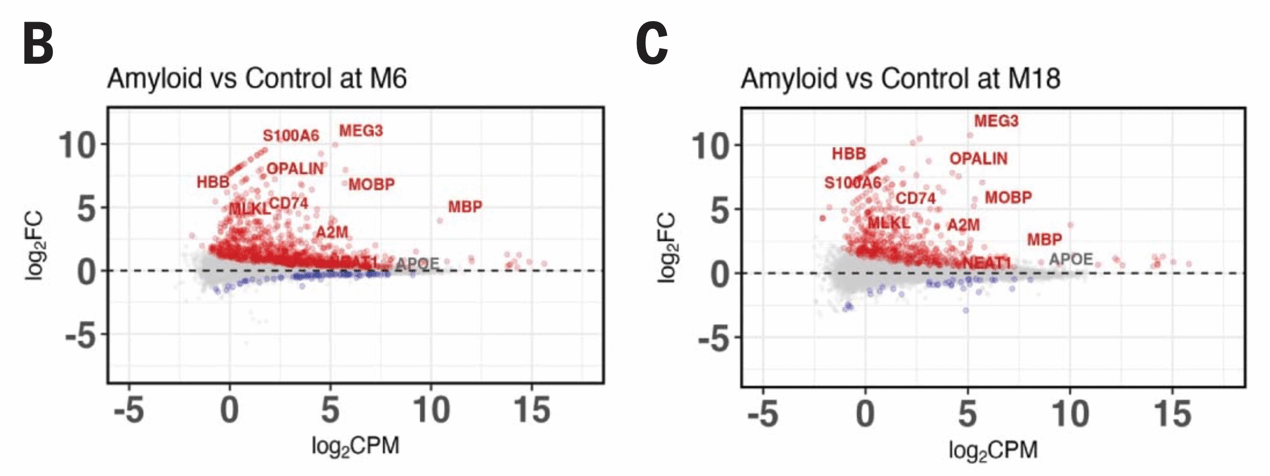
图5 MEG3表达量显著上调
3、MEG3调节人源神经元的坏死途径
接着,研究者进行了体外实验验证MEG3的作用。他们使用慢病毒在H9衍生的成熟皮层神经元中表达MEG3,与对照组相比,表达MEG3的细胞在第9天时活力显著降低,免疫组织化学分析显示存在活化的坏死标记物pRIPK1(Ser166)、pRIPK3(Ser227)和pMLKL(Ser358)。

图6 体外过表达MEG3可以诱导人类神经元的坏死性凋亡
且MEG3过表达诱导的神经元程序性坏死,可以通过CRISPR敲除程序性坏死相关蛋白RIPK1、RIPK3和MLKL,或者使用程序性坏死抑制剂ponatinib、dabrafenib、necrosulfonamide来挽救。
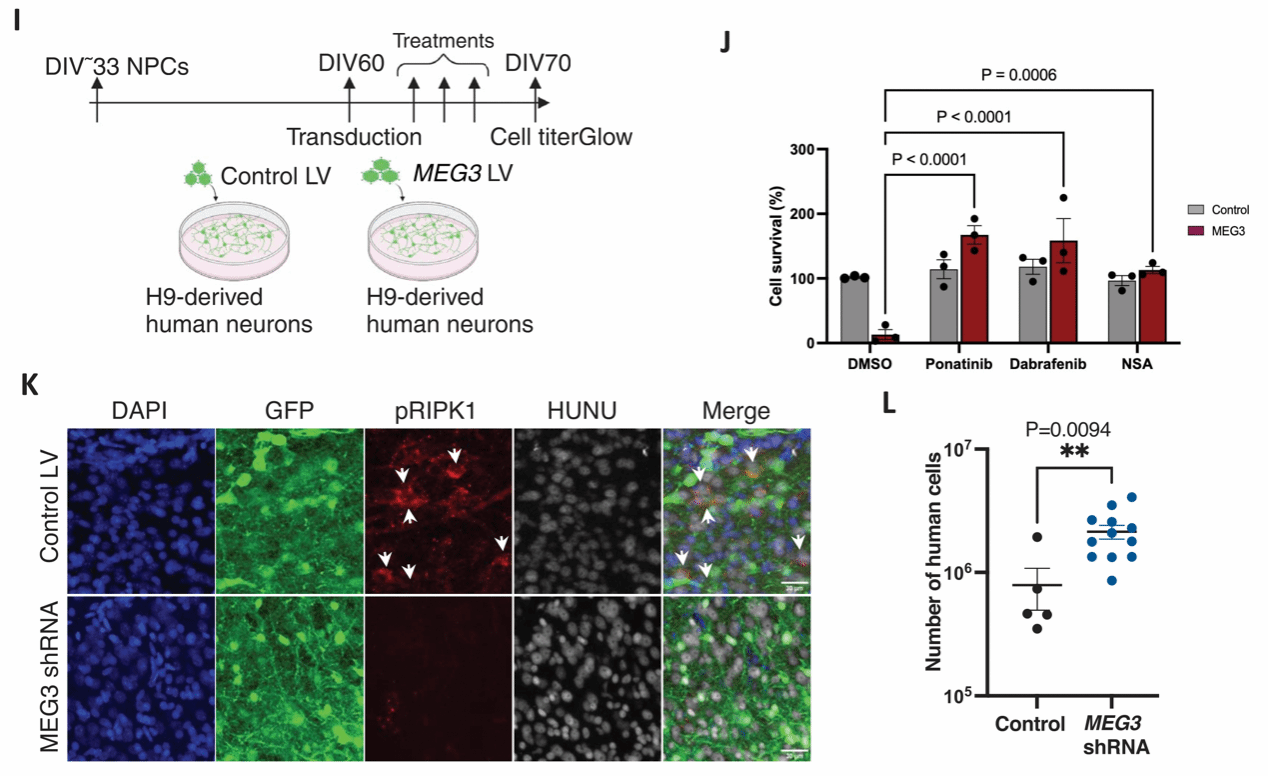
图7 药理学或遗传学手段下调MEG3的表达可以挽救MEG3诱导的神经元缺失
4、抑制坏死可以阻止异种移植人源神经元丢失
进一步,研究者企图探究MEG3介导的坏死途径是否也在体内介导了异种移植人源神经元的丢失。研究者在移植后第2个月至第6个月,给异种移植小鼠口服坏死性凋亡激酶抑制剂ponatinib和dabrafenib进行治疗(ponatinib是是RIPK1和RIPK3抑制剂,已被FDA批准用于治疗急性淋巴细胞白血病和慢性髓细胞白血病;dabrafenib是一种特异性更强的RIPK3抑制剂)。研究发现,移植后第6个月,两组治疗组小鼠的程序性坏死蛋白(pRIPK1、pRIPK3和pMLKL)水平相较未治疗组均大幅降低,dabrafenib组小鼠神经元死亡也显著减少。
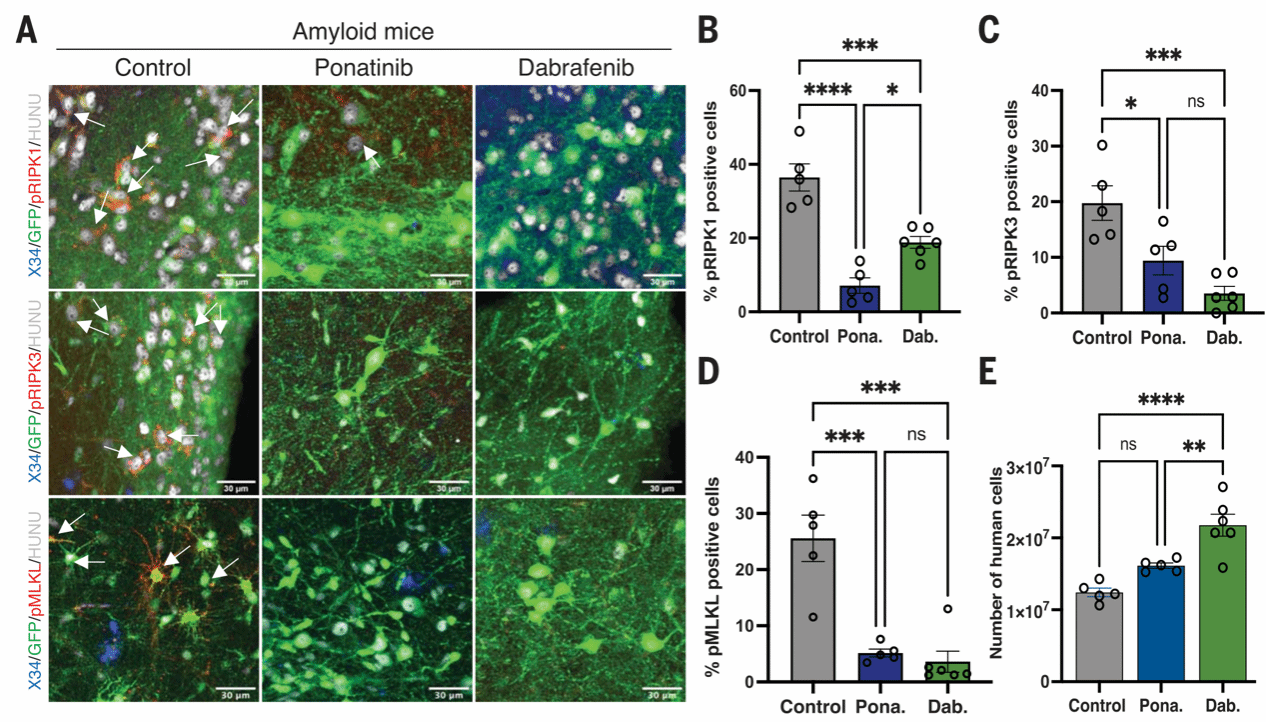
图8 抑制坏死可防止人源神经元在体内丢失
综上,这项发表于 Science 的最新研究,通过将人类或小鼠神经元移植进淀粉样蛋白小鼠模型的大脑中,发现只有人类神经元表现出严重的阿尔茨海默病病理并发生坏死性凋亡,而MEG3的急剧上调是其关键因素。该研究揭示了人类神经元对AD的独特易感性,也提示我们或许可以对小鼠的内源性神经元进行基因改造,使得小鼠的MEG3也能够响应Aβ病理,从而开发出新的AD小鼠动物模型。
原文链接:
https://www.science.org/doi/10.1126/science.abp9556
参考文献:
[1] HOLTZMAN D M, MORRIS J C, GOATE A M. A new mouse model to study late-onset Alzheimer’s disease [J]. Sci Transl Med, 2011, 3: 77sr1.
[2] SCHELTENS P, DE STROOPER B, KIVIPELTO M, et al. Alzheimer's disease [J]. Lancet, 2021, 397(10284): 1577-90.
[3] KOSEL F, PELLEY J M S, FRANKLIN T B. Behavioural and psychological symptoms of dementia in mouse models of Alzheimer's disease-related pathology [J]. Neurosci Biobehav Rev, 2020, 112: 634-47.
[4] CHEN X, HOLTZMAN D M. Emerging roles of innate and adaptive immunity in Alzheimer's disease [J]. Immunity, 2022, 55(12): 2236-54.
[5] GOTZ J, ITTNER L M. Animal models of Alzheimer's disease and frontotemporal dementia [J]. Nat Rev Neurosci, 2008, 9(7): 532-44.
[6] HE Z, GUO J L, MCBRIDE J D, et al. Amyloid-beta plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation [J]. Nat Med, 2018, 24(1): 29-38.
[7] MATTSSON-CARLGREN N, ANDERSSON E, JANELIDZE S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease [J]. Science Advances, 2020, 6(16): eaaz2387.




 地址:上海市徐汇区医学院路138号
地址:上海市徐汇区医学院路138号  邮编:200032
邮编:200032  电话/传真:021-54237056
电话/传真:021-54237056  邮箱:itbr@fudan.edu.cn
邮箱:itbr@fudan.edu.cn 