神经酰胺作为主要复合鞘脂的前体,是细胞膜结构的组成部分,并在包括细胞生长、分化、增殖和凋亡的各种细胞过程中发挥调节作用。在过去的三十年中,神经酰胺被认为是代谢功能障碍的关键驱动因素,并且发现人体内高内源性浓度的神经酰胺脂质与2型糖尿病(T2 D)、肥胖症、肝脂肪变性、动脉粥样硬化和其他心血管疾病密切相关【1-5】。例如,与正常饮食(NCD)相比,12周或16周高脂饮食(HFD)治疗可诱导小鼠皮下白色脂肪组织(scWAT)和棕色脂肪组织(BAT)中神经酰胺水平的增加【6-8】。此外,病理性增加的长链神经酰胺水平被发现与小鼠或人类患者的非酒精性脂肪肝(NAFLD)和糖尿病视网膜病变有关。
来自山东大学基础医学院孙金鹏团队,联合北京大学医学部基础医学院姜长涛和孔炜教授、山东大学于晓团队合作鉴定出FPR2是一种跨膜受体,能够特异性结合长链神经酰胺(C14-C20)。在棕色和米色脂肪细胞中,C16:0神经酰胺与FPR2结合会通过Gi-环AMP信号通路抑制产热,而在FPR2缺失的情况下,这一效应被逆转。研究者解析了FPR2分别与C16:0、C18:0和C20:0神经酰胺结合并与Gi三聚体复合的三种冷冻电镜结构。神经酰胺的疏水尾深入嵌入到正构配体口袋内,该口袋的构象可塑性有限。对结构相近的受体(如FPR1或FPR3)的神经酰胺结合基序进行修饰,可以使其从无活性转变为活性神经酰胺受体。本研究结果提供了FPR2介导的脂肪细胞产热的结构基础。该研究与2025年3月发表于Science杂志上。
1.研究团队在体外实验中发现,C16:0神经酰胺短时间刺激(15分钟)会降低棕色脂肪组织、米色脂肪组织及其相应细胞内的cAMP含量,提示可能存在一种Gi偶联的GPCR受体介导神经酰胺对脂肪组织的快速调控作用。随后,团队利用GloSensor-cAMP方法筛选棕色脂肪中表达量前60位的GPCR,发现C16:0神经酰胺可激活FPR2受体导致cAMP含量下降。通过BRET-G蛋白解离实验进一步验证,发现C16:0神经酰胺通过激活FPR2的Gi1和Gi2信号通路发挥作用,最终确定FPR2是脂肪细胞中Gi偶联的神经酰胺受体。
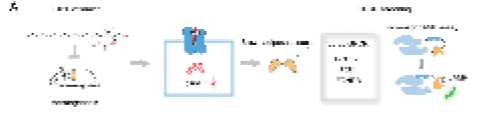
图1 用于筛选C16:0神经酰胺传感GPCR的策略示意图
2.为了验证神经酰胺与FPR2之间是否存在直接相互作用,研究团队采用了放射性同位素配体结合和FlAsH-BRET传感器方法。实验结果表明,神经酰胺与FPR2之间存在直接相互作用。在放射性同位素配体结合实验中,C16:0神经酰胺在棕色脂肪组织和皮下白色脂肪组织中的结合亲和力(Ki值),分别为600.5 ± 38.9 nM和190.7 ± 25.7 nM。FlAsH-BRET传感器实验进一步显示,C16:0神经酰胺与FPR2结合可触发受体特异性的胞外构象变化,表现为FPR2的N端向ECL1和ECL2区域靠近 。
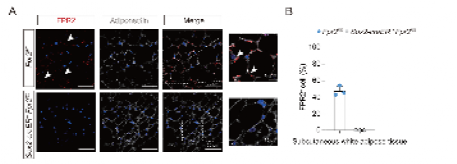
图2 小鼠的scWAT脂肪细胞中FPR 2免疫染色水平
3.为了明确FPR2是否介导C16:0神经酰胺对脂肪产热的抑制作用,研究团队构建了冷刺激产热模型。发现在Ucp1-Cre+/−Fpr2fl/fl和Adipoq-Cre+/−Fpr2fl/fl两种敲除小鼠中,或通过使用Fpr2的拮抗剂WRTW4干预后,在不影响小鼠饮食和运动的情况下,均显著减轻神经酰胺对脂肪产热的抑制作用,小鼠表现为能量代谢和氧消耗量显著增加、体温升高,且脂肪组织中产热基因表达及UCP1蛋白水平上调。上述结果表明,在生理冷刺激条件下,C16:0神经酰胺通过激活脂肪细胞的FPR2受体抑制产热。
4.脂肪产热是抵抗肥胖的关键机制。为了探究神经酰胺在肥胖中的作用,研究团队分别构建了高脂饮食诱导的适应性产热模型(高脂饮食2周)和肥胖模型(高脂8周)。在适应性产热模型中,Adipoq-Cre+/−Fpr2fl/fl小鼠的脂肪产热能力增强,同时其糖耐量受损和胰岛素抵抗显著改善。在肥胖模型中,与对照组相比,Adipoq-Cre+/−Fpr2fl/fl小鼠的体重增长显著减缓,且肝脏组织、皮下白色脂肪组织、内脏白色脂肪组织和棕色脂肪组织的重量及甘油三酯水平显著下降。上述结果表明,靶向FPR2受体可能通过调控脂肪产热影响肥胖及相关代谢紊乱的进程。

图4 神经酰胺处理小鼠能量消耗值
5.最后,研究人员解析了神经酰胺-FPR2-Gi复合物的三维结构。发现神经酰胺的鞘链深埋于FPR2正构结合口袋底部,而脂肪酸链则在口袋上方呈现特征性折叠构象(“p”或“S”形)。这种独特的结合模式阐释了FPR2对神经酰胺的选择性识别机制—过长的脂肪酸链和含双键脂肪酸链的神经酰胺会因口袋可容纳空间不足导致结合能显著降低。基于此发现,研究人员通过系统性的定点突变策略,成功将FPR1/FPR3(同家族非神经酰胺敏感受体)改造为神经酰胺响应型受体。这不仅验证了关键功能基序的分子逻辑,更为GPCR家族的功能重编程提供了范式参考。

图5 神经酰胺-FPR2-Gi复合物的整体结构
总结
本项研究确定FPR2作为启动Gi信号传导的内源性神经酰胺受体,其介导神经酰胺对脂肪细胞产热的抑制作用。与不同神经酰胺复合的FPR 2的药理学表征和Cryo-EM结构提供了FPR2作为神经酰胺受体的作用的具体证据。在哺乳动物中,有数百种不同种类的神经酰胺,目前的研究仅集中在脂肪组织中限制的16:0神经酰胺受体筛选。本项研究的平行研究中,作者发现Gq偶联神经酰胺受体CYSLTR 2和P2RY6参与动脉粥样硬化的恶化。基于这项工作,作者预计,其他膜神经酰胺受体存在识别特定的神经酰胺和参与病理生理过程。
参考文献
【1】 S. A. Summers, B. Chaurasia, W. L. Holland, Metabolic Messengers: Ceramides. Nat. Metab. 1, 1051–1058 (2019).
【2】 Y. A. Hannun, L. M. Obeid, Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 (2008).
【3】 R. H. Choi, S. M. Tatum, J. D. Symons, S. A. Summers, W. L. Holland, Ceramides and other sphingolipids as drivers of cardiovascular disease. Nat. Rev. Cardiol. 18, 701–711 (2021).
【4】 S. M. Turpin-Nolan, J. C. Brüning, The role of ceramides in metabolic disorders: When size and localization matters. Nat. Rev. Endocrinol. 16, 224–233 (2020).
【5】W. L. Holland, J. T. Brozinick, L.-P. Wang, E. D. Hawkins, K. M. Sargent, Y. Liu, K. Narra, K. L. Hoehn, T. A. Knotts, A. Siesky, D. H. Nelson, S. K. Karathanasis, G. K. Fontenot, M. J. Birnbaum, S. A. Summers, Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 5, 167–179 (2007).
【6】S. M. Turpin, H. T. Nicholls, D. M. Willmes, A. Mourier, S. Brodesser, C. M. Wunderlich, J. Mauer, E. Xu, P. Hammerschmidt, H. S. Brönneke, A. Trifunovic, G. LoSasso, F. T. Wunderlich, J.-W. Kornfeld, M. Blüher, M. Krönke, J. C. Brüning, Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 20, 678–686 (2014).
【7】C. Shah, G. Yang, I. Lee, J. Bielawski, Y. A. Hannun, F. Samad, Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J. Biol. Chem. 283, 13538–13548 (2008).
【8】H. Liu, P. Wang, F. Xu, Q. Nie, S. Yan, Z. Zhang, Y. Zhang, C. Jiang, X. Qin, Y. Pang, The Hydrophilic Metabolite UMP Alleviates Obesity Traits through a HIF2αACER2-Ceramide Signaling Axis. Adv. Sci. (Weinh.) 11, e2309525 (2024).
本文作者:杨涵婷组王馨敏




 地址:上海市徐汇区医学院路138号
地址:上海市徐汇区医学院路138号  邮编:200032
邮编:200032  电话/传真:021-54237056
电话/传真:021-54237056  邮箱:itbr@fudan.edu.cn
邮箱:itbr@fudan.edu.cn 