慢性疼痛给个人和经济带来了巨大的负担,对全球30%以上的人产生影响[1]。目前针对慢性疼痛的非阿片类药物疗效较低,而阿片类药物具有滥用倾向且过量使用时可致死[2],因此开发更安全、更有效的慢性疼痛治疗药物具有重要意义。
大麻素1型受体(Cannabinoid receptor type 1,CB1)是一个备受关注的非阿片类靶点。其广泛表达在疼痛神经通路中,对应的激动剂在几种不同的动物疼痛模型中都表现出镇痛作用[3, 4]。最近,多种高效合成大麻素受体激动剂(synthetic cannabinoid receptor agonists,SCRAs)已被发现,而使用SCRAs治疗慢性疼痛有两个关键障碍:1)SCRAs会激活中枢神经系统的CB1受体,产生不良的副作用;2)SCRAs会促进CB1受体与β-arrestin的相互作用,进而导致耐受性。

(文章标题和作者)
来自美国华盛顿大学医学院的Susruta Majumdar和Robert W. Gereau IV以及斯坦福大学的Ron O. Dror和Kaavya Krishna Kumar团队合作通过基于结构的理性设计策略开发了新一代CB1受体激动剂。该激动剂不仅具有外周限制特性,而且能够选择性抑制β-arrestin信号传导。相关研究“A cryptic pocket in CB1 drives peripheral and functional selectivity”于2025年3月5日发表在《Nature》杂志上。
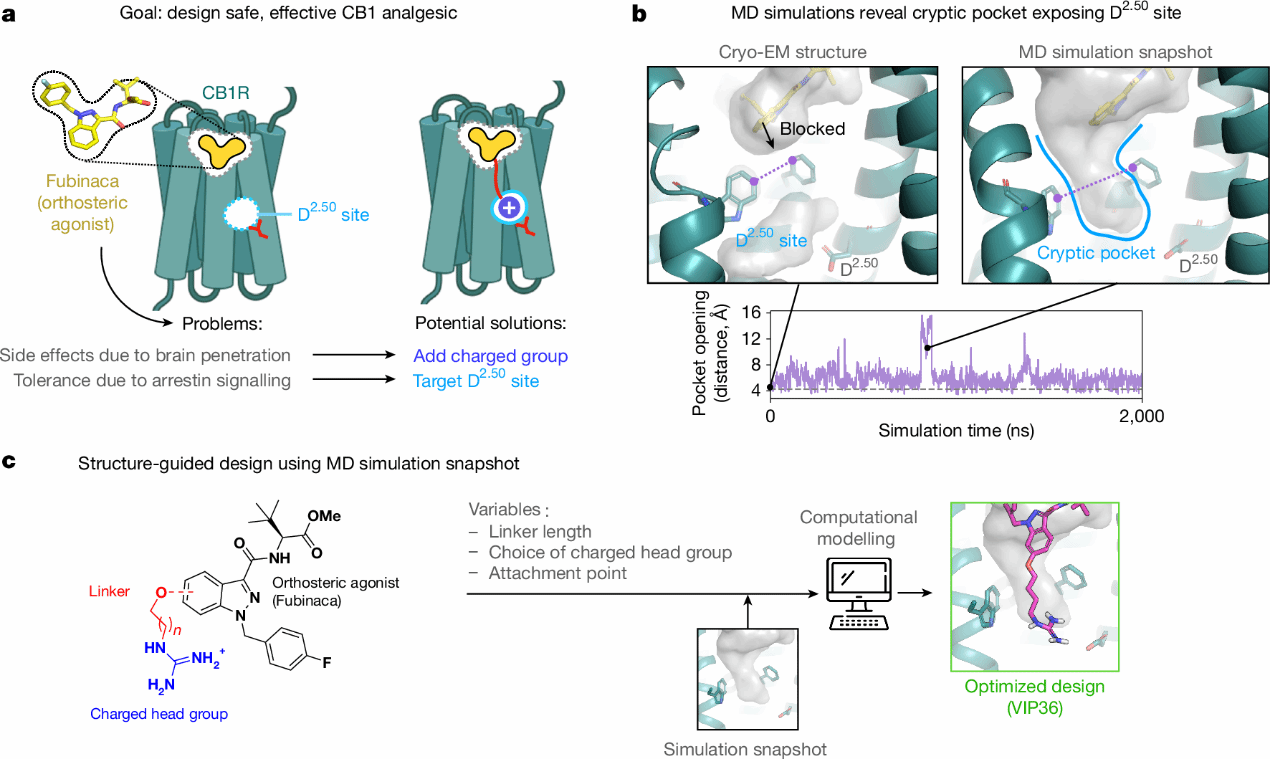
图1 基于结构的CB1靶向镇痛药的设计
为开发能够克服包括中枢神经系统副作用和耐受性在内的两个关键障碍的激动剂,研究人员尝试引入针对CB1的D2.50位点的带电官能团。前期研究表明,增加配体电荷会降低对中枢神经系统的透入,在GPCR如μ-阿片受体(μOR)核心内靶向D2.50位点能降低β-arrestin信号传导效率。然而,在所有已解析的CB1结构中,庞大的芳香族残基F2003.36和W3566.48(称为构象转换残基)阻断了从正构结合口袋进入D2.50位点的通路。此外,与μOR和许多其他GPCR相反,CB1的药理学实验没有提供与D2.50相邻的Na+结合位点的证据,这进一步引发了对D2.50位点可及性的质疑。
为确认D2.50位点是否可以在其他受体构象中可及,研究人员对与高效激动剂MDMB-Fubinaca (FUB)结合的CB1进行了分子动力学模拟。模拟过程捕捉到了从正位结合口袋延伸到D2.50位点的瞬态隐蔽口袋。这一先前未知的隐蔽口袋开放概率低,仅占模拟时间的8%,并且需要构象转换残基的分离,特别是W3566.48的旋转。激动剂可能在隐蔽口袋短暂开放时进入其中,并形成有利的相互作用,进一步稳定其开放构象。因此,模拟结果提供了一个可访问D2.50位点的受体构象,为基于结构的药物设计提供了可能性。
基于上述隐蔽口袋的模拟结果,研究人员以FUB骨架为起点,引入了带正电荷头基的柔性linker,设计了一系列预计能够延伸至D2.50位点的FUB类似物。通过预测获得了VIP36,其结构通过四碳O-烷基linker将胍基与FUB的吲唑环相连,能够与D2.50位点形成紧密相互作用。
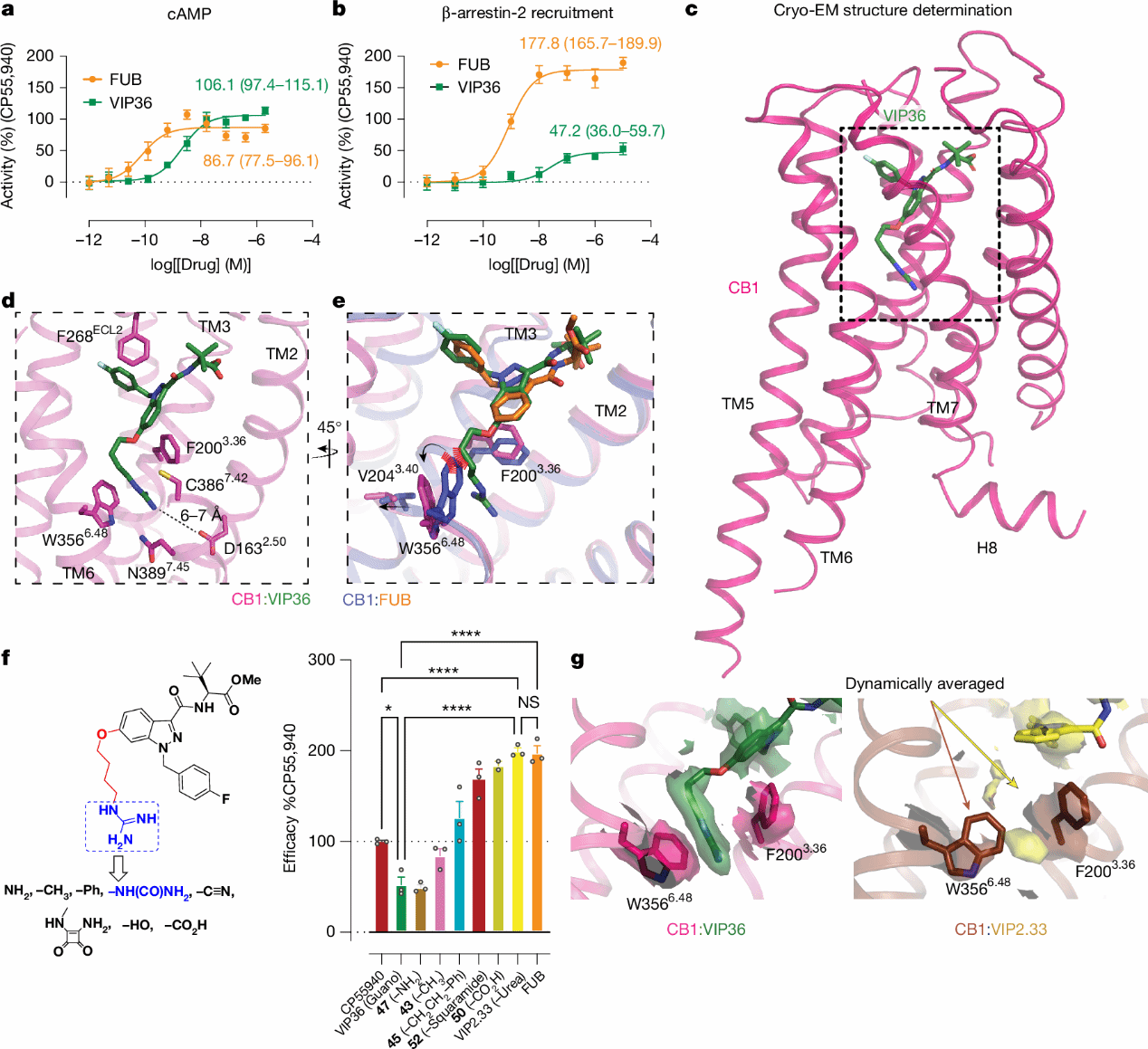
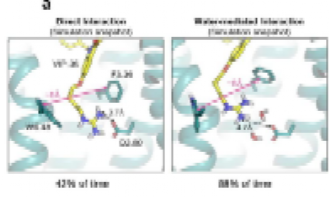
图2 VIP36-CB1的冷冻电镜结构
为研究VIP36与CB1受体的相互作用并验证其结合机制,研究人员解析了VIP36与Gi1偶联的CB1复合物的冷冻电镜结构,分辨率为2.86 Å。VIP36在表征G蛋白活性的cAMP测定和基于BRET的β-arrestin-2募集测定中显示出符合预期的G蛋白信号传导偏向性。其FUB部分与CB1受体之间存在一系列保守相互作用,包括第二胞外环(ECL2)的F268ECL2残基直接堆积,以及与关键构象转换残基F2003.36形成π-π相互作用。
值得注意的是,冷冻电镜密度图明确显示VIP36中的胍基位于预测的隐蔽口袋中。“常规活性”构象中的W3566.48与VIP36中的胍基linker不兼容,因此W3566.48向外移动到初始分子动力学模拟所预测的构象,导致其“活性-开放”构象,并伴随TM3螺旋的V2043.40残基外移以适应新的旋转状态。
VIP36的胍基在冷冻电镜结构中未直接接触D2.50位点,而是与构象转换残基形成阳离子-π相互作用(与W3566.48和F2003.36的距离分别为4.0 Å和4.4 Å),并与N3897.45形成强氢键。D2.50位点与胍基距离6-7 Å,排除了二者具有典型盐桥相互作用的可能性。这一现象与在μOR中的研究显著不同,后者设计的双位点配体与D2.50位点存在清晰的离子相互作用。将VIP36-CB1的冷冻电镜结构嵌入水合脂质双层中进行了分子动力学模拟,能观察到VIP36与D2.50位点间存在长程水介导的相互作用,而有时则是短程(<3.5 Å)电荷-电荷相互作用。


图3 CB1受体中配体信号传导偏向性的分子机制
为揭示VIP36介导的G蛋白信号传导偏向性的确切分子机制,研究人员解析了结合VIP2.33的CB1冷冻电镜结构。VIP2.33是用氧取代了VIP36中的亚胺氮后获得的带尿素头基的FUB类似物。这一单原子取代导致arrestin蛋白募集增加约四倍,类似于原始FUB模板。冷冻电镜结构比较发现,CB1与VIP36结合后,相邻的跨膜螺旋7(TM7)稳定在一种构象,而与VIP2.33结合后,TM7优先采用替代的旋转构象。而TM7构象已被证明调节其他GPCRs中的偏向性信号传导。
进一步的分子动力学模拟表明VIP36能直接与D2.50和TM7相互作用,建立一个显著影响TM7构象的接触网络。在结构-活性关系(SAR)分析中,带正电荷的官能团对G蛋白信号传导偏向性的重要性,支持了在配体和D2.50之间观察到的离子相互作用对于形成这种相互作用网络是至关重要的。因此,综合上述结果,提出了一个结构机制:配体、D2.50和TM7之间的相互作用最终控制细胞内受体构象以引起信号偏差。
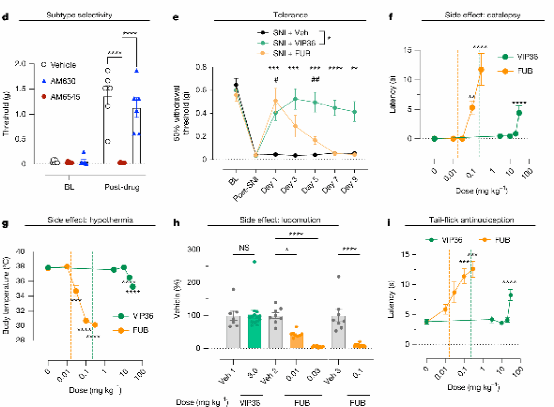
图4 VIP36在动物模型中具有镇痛作用,减少不良反应和耐受性
为验证VIP36的外周限制特性和有限的耐受性,研究人员通过三种不同的体内疼痛模型评估VIP36的镇痛效果,包括CFA、SNI和NTG。结果表明,VIP36在炎症、神经病理和偏头痛模型中表现出镇痛活性,且呈现剂量依赖性和时间依赖性。
其中,对SNI模型小鼠腹腔注射VIP36(1 mg/kg),每日两次,持续9天,逆转了SNI诱导的异常疼痛,且这一效果在测试期间持续存在。相比之下,FUB在第3天开始出现耐受性迹象。
对中枢CB1受体参与而产生的包括低温、昏厥、运动活动改变和抗伤害感受在内的反应检测,表明VIP36介导的镇痛效果和中枢系统的副作用之间的剂量间隔为100倍。
综上所述,该研究设计了一种外周选择性CB1激动剂VIP36,其β-arrestin募集降低,可以巩固高效SCRAs的镇痛作用,同时避免由中枢活性介导的不良反应和arrestin募集产生的耐受性。在炎症、神经病理和偏头痛小鼠模型中,VIP36均能产生镇痛作用。这些结果表明,靶向GPCR的隐蔽口袋能够增强外周选择性和信号偏向性,同时促进体内药理学应用和不良反应最小化。这对于慢性疼痛的治疗有实质性的意义,还可能革新其他GPCR靶向药物的设计。
[1] COHEN S P, VASE L, HOOTEN W M. Chronic pain: an update on burden, best practices, and new advances [J]. Lancet, 2021, 397(10289): 2082-2097.
[2] VOLKOW N D, BLANCO C. The changing opioid crisis: development, challenges and opportunities [J]. Mol Psychiatry, 2021, 26(1): 218-233.
[3] WOODHAMS S G, CHAPMAN V, FINN D P, et al. The cannabinoid system and pain [J]. Neuropharmacology, 2017, 124: 105-120.
[4] PERTWEE R G. Pharmacology of cannabinoid CB1 and CB2 receptors [J]. Pharmacol Ther, 1997, 74(2): 129-180.
本文作者:张凯华组周琳




 地址:上海市徐汇区医学院路138号
地址:上海市徐汇区医学院路138号  邮编:200032
邮编:200032  电话/传真:021-54237056
电话/传真:021-54237056  邮箱:itbr@fudan.edu.cn
邮箱:itbr@fudan.edu.cn 