On November 29, 2022, a collaborative study led by Prof. Wei-Guang Li from our institute, in cooperation with Prof. Fei Li's team from Xinhua Hospital affiliated with Shanghai Jiao Tong University School of Medicine, Prof. Xiao-Yong Zhang's group from the Institute of Brain-inspired Intelligence Science and Technology at Fudan University, and Prof. Bing Mei's lab from East China Normal University, was published online in the journal Cell Reports (Cell Press), titled "Social deficits via dysregulated Rac1-dependent excitability control of prefrontal cortical neurons and increased GABA/glutamate ratios". Employing a multidisciplinary approach including mouse models of autism spectrum disorder (ASD), behavioral assays, magnetic resonance spectroscopy (MRS), optogenetics, chemogenetics, electrophysiology, molecular genetics, and cellular biology, this study addresses the clinical complexity and heterogeneity of social deficits in autism. By elucidating how bidirectional dysregulation of Rac1—a key regulator of synaptic cytoskeletal dynamics—in distinct cell types within the medial prefrontal cortex (mPFC) leads to neuronal excitability changes and neurotransmitter imbalances closely linked to abnormal social behaviors, the researchers proposed a novel classification framework for autism social deficits based on synaptic and cellular biological characteristics. The findings provide important theoretical insights for understanding common pathological mechanisms of ASD social dysfunction and lay a foundation for developing biology-informed diagnostic and therapeutic strategies.

Autism spectrum disorder is an early-onset neurodevelopmental disorder characterized by severe social impairments as a core symptom. Its incidence is steadily increasing worldwide. Accumulating evidence indicates that a combination of environmental and genetic factors significantly increases susceptibility to ASD. Due to the complexity of ASD etiology and the remarkable heterogeneity of clinical presentations, there remains a significant lack of effective treatments, posing major clinical challenges. Identifying common neural mechanisms underlying social deficits and developing biologically grounded classification systems are thus critical steps toward precise therapeutic interventions.
Synapses, the fundamental units for neuronal communication, exist as either excitatory or inhibitory types. An imbalance between synaptic excitation and inhibition (E/I imbalance) is widely recognized as a key pathological mechanism underlying autism and other neuropsychiatric disorders. However, how diverse social deficits in autism correlate precisely with specific types of synaptic imbalance remains unclear. In this study, researchers focused on Rac1, a small GTPase critical for regulating synaptic cytoskeletal organization and plasticity. Rac1 cycles between active (GTP-bound) and inactive (GDP-bound) states, regulating actin cytoskeletal dynamics and synapse remodeling. Clinical evidence has shown that both gain- and loss-of-function RAC1 mutations are strongly linked to autism risk. Animal studies have demonstrated altered Rac1 activity in different autism mouse models such as fragile X syndrome mice (Fmr1 KO; increased Rac1 activity) and Shank3 C-terminal deletion mice (Shank3+/ΔC; decreased Rac1 activity). However, whether and how Rac1 dysregulation—either increased or decreased—contributes directly to social deficits, and whether these effects depend on specific cell-type regulation, remained unknown.
To establish a causal link between Rac1 activity and social deficits, the researchers employed viral stereotactic manipulations combined with behavioral assays in mice. They found that bidirectional manipulation (both increases and decreases) of Rac1 activity within the mPFC, a critical region implicated in social behavior, impaired social interactions. Using magnetic resonance spectroscopy (MRS), the researchers further revealed distinct neurotransmitter profiles: increased Rac1 activity reduced glutamate without affecting GABA levels, whereas decreased Rac1 activity elevated GABA without altering glutamate levels. Both manipulations ultimately resulted in increased inhibitory/excitatory (GABA/glutamate) ratios in the mPFC.
Given the cellular diversity within the mPFC, comprising excitatory glutamatergic neurons and inhibitory GABAergic interneurons such as parvalbumin (PV)-positive cells, researchers performed cell-type-specific Rac1 manipulations. They discovered that upregulating Rac1 specifically in excitatory glutamatergic neurons reduced neuronal excitability and glutamate levels, whereas downregulating Rac1 selectively in PV-positive inhibitory neurons enhanced neuronal excitability and GABA release. Both manipulations impaired social novelty recognition behavior (a measure of social memory). Remarkably, chemogenetic or optogenetic activation of glutamatergic neurons rescued social deficits induced by Rac1 upregulation, while chemogenetic inhibition of PV neurons reversed deficits caused by Rac1 downregulation. Conversely, manipulating Rac1 in the opposite direction in these neurons did not affect social behavior.
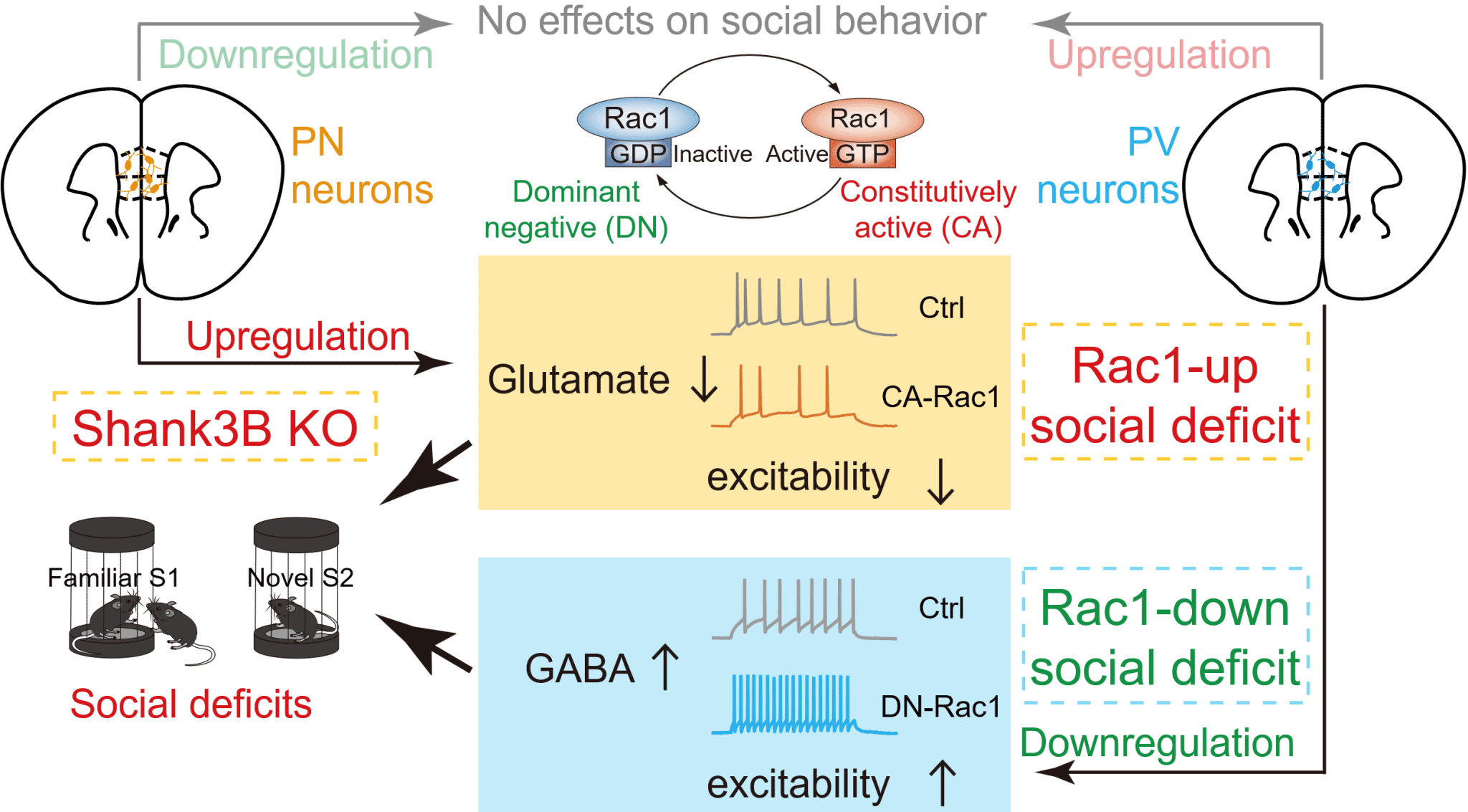
Figure. Schematic illustrating how bidirectional alterations of Rac1 activity in distinct neuronal types within the mPFC lead to social deficits.
Based on these results, the team proposed a novel classification scheme for autism-related social deficits, defined by synaptic cellular biology: the "Rac1-up subtype", characterized by elevated Rac1 activity primarily affecting glutamatergic neurons (resulting in reduced glutamate levels), and the "Rac1-down subtype", marked by reduced Rac1 activity mainly affecting inhibitory interneurons (resulting in elevated GABA levels). Both subtypes converge on increased inhibitory/excitatory ratios within the mPFC (see schematic figure). Interestingly, "Rac1-down subtype" deficits involve generalized impairments in novelty recognition, while "Rac1-up subtype" deficits selectively affect social novelty recognition.
To validate this framework, researchers examined a distinct autism model mouse, Shank3B KO (lacking the PDZ domain of the Shank3 gene), and found impaired social memory accompanied by elevated Rac1 activity, decreased glutamatergic neuronal excitability, and reduced glutamate levels in the mPFC, closely resembling the Rac1-up subtype. In contrast, this phenotype was opposite to the Rac1-down subtype seen in Shank3+/ΔC mice. Targeted inhibition of Rac1 activity or enhancement of neuronal excitability specifically in mPFC glutamatergic neurons effectively alleviated social deficits in Shank3B KO mice.
Authorship and Acknowledgments:
Dr. Bingke Ma, a doctoral candidate from East China Normal University, is the first author of the study. Prof. Fei Li (Xinhua Hospital affiliated with Shanghai Jiao Tong University School of Medicine), Prof. Xiao-Yong Zhang (Institute of Brain-inspired Intelligence Science and Technology at Fudan University), Prof. Bing Mei (East China Normal University), and Prof. Wei-Guang Li (our institute) are the co-corresponding authors.
This research was supported by the National Natural Science Foundation of China, Shanghai Science and Technology Commission, Shanghai Municipal Health Commission, and Shanghai Municipal Education Commission. Collaborative support was also provided by Prof. Xiaobing Yuan and Prof. Dongmin Yin from East China Normal University, Prof. Juehua Yu from the First Affiliated Hospital of Kunming Medical University, and Prof. Zhonghua Lu from the Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences.
Link to original paper: https://doi.org/10.1016/j.celrep.2022.111722






 Address:No.138, medical college road
Address:No.138, medical college road  Postcode:200032
Postcode:200032  Tel/Fax:021-54237056
Tel/Fax:021-54237056 Emial:itbr@fudan.edu.cn
Emial:itbr@fudan.edu.cn