On June 21, 2024, a collaborative study led by Prof. Wei-Guang Li from our institute and Prof. Tian-Le Xu from Shanghai Jiao Tong University School of Medicine was published online in Nature Communications, entitled "Sensory ASIC3 channel exacerbates psoriatic inflammation via a neurogenic pathway in female mice". Using a mouse model of psoriasis—a chronic inflammatory skin disease—and employing multidisciplinary approaches including genetics, electrophysiology, metabolomics, and electrochemistry, this study reveals a novel neuroimmune mechanism whereby acid-sensing ion channel type 3 (ASIC3), expressed on peripheral sensory neurons, senses changes in the skin's metabolic microenvironment, thus exacerbating inflammation and lesion formation. This discovery provides a new therapeutic perspective for refractory skin diseases.

Psoriasis, commonly known as a chronic and recurrent inflammatory skin disorder, affects millions worldwide. It manifests as scaly erythematous plaques that significantly impair quality of life. Psoriasis pathogenesis involves abnormal immune responses and excessive proliferation and aberrant differentiation of keratinocytes. Recent research has increasingly highlighted the critical role of peripheral sensory neurons innervating the skin in psoriasis, particularly interactions between nociceptor nerve endings and dermal dendritic cells (DCs), which coordinate the hallmark Th17-type immune response. Evidence suggests that nociceptive nerve terminals release calcitonin gene-related peptide (CGRP), stimulating dendritic cell activation through contact-dependent calcium influx, membrane depolarization, and secretion of chemokine CCL2, thus orchestrating local inflammation and adaptive immunity. Despite progress in understanding nociceptor-immune interactions in psoriatic inflammation, the molecular mechanisms coordinating immune responses within the skin barrier remain unclear.
Using an imiquimod (IMQ)-induced mouse model of psoriasis, the researchers applied electrochemical analyses combined with lipid metabolomics to characterize the inflammatory microenvironment within psoriatic lesions and elucidate the underlying neuroimmune regulation of chronic skin inflammation. The team found time-dependent acidosis within psoriatic-like skin lesions, accompanied by dysregulated lipid metabolism, notably increased lysophosphatidylcholine (LPC) levels. Intriguingly, both tissue acidosis and elevated LPC can activate ion channels on peripheral sensory neurons that sense extracellular pH, especially ASIC3. Upon activation, ASIC3 mediates sodium ion influx, thus regulating neuronal excitability and sensory functions such as pain and itch. However, the precise role of ASIC3 in psoriatic inflammatory processes remained unexplored. Researchers thus hypothesized that ASIC3 senses local increases in protons (H+) and LPC, influencing the progression of psoriasis-like pathology.
Employing global Asic3 knockout mice (Asic3 KO) and conditional knockout mice specifically lacking ASIC3 in peripheral nociceptors (NaV1.8Cre::Asic3flox/flox), the researchers demonstrated that ASIC3, particularly on peripheral nociceptors, is essential for psoriasis lesion formation. ASIC3 deficiency significantly ameliorated IMQ-induced psoriatic symptoms. Additional genetic manipulations and pharmacological interventions confirmed the critical role of nociceptor-expressed ASIC3 in psoriasis pathology. Moreover, psoriatic mice exhibited abnormal sensory nerve sprouting, with significantly increased CGRP-positive nerve endings in lesional skin. In cultured mouse dorsal root ganglion (DRG) neurons, ASIC3 activation was shown to enhance CGRP release.
Co-culture experiments further revealed that ASIC3-driven CGRP release stimulated dendritic cells to produce interleukin-23 (IL-23), subsequently promoting keratinocyte proliferation and contributing to psoriasis pathology. Interestingly, direct IL-23 administration triggered psoriasis-like lesions even in ASIC3-deficient mice, suggesting that a sensory-neural pathway involving ASIC3→CGRP→dendritic cells→IL-23 constitutes a novel neuroimmune regulatory axis in psoriasis. Consistent with this, treatment with botulinum neurotoxin A (BoNT/A) or a CGRP antagonist alleviated psoriatic inflammation comparably to ASIC3 knockout. Conversely, supplementation of CGRP in the skin of Asic3 KO mice restored inflammatory responses. These findings not only reveal a novel mechanism of ASIC3-mediated neurogenic inflammation in psoriasis but also provide potential targets for therapeutic interventions against neurogenic inflammation (Fig. 1).
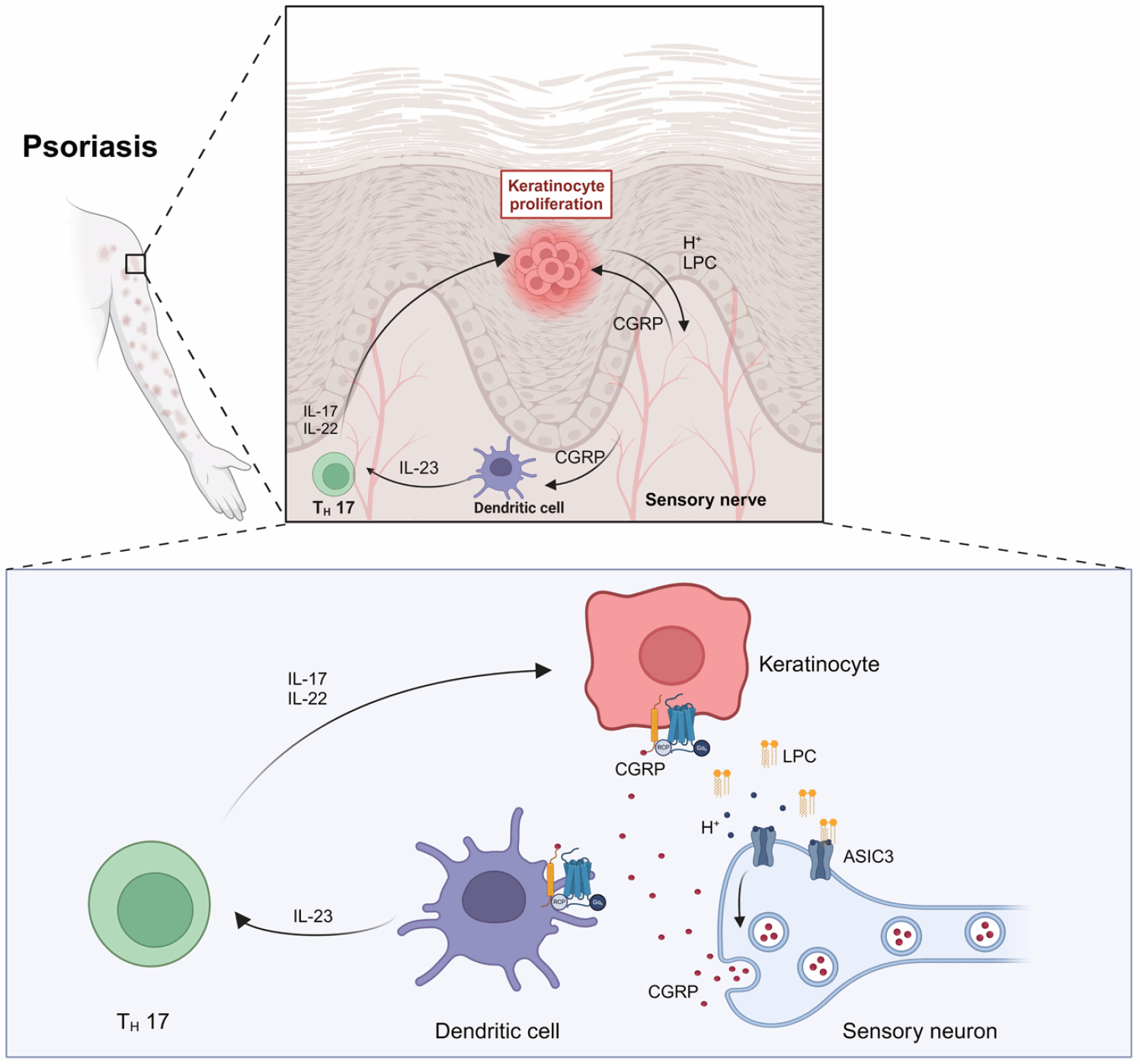
Figure 1. Working model illustrating how ASIC3 channels on sensory neurons exacerbate psoriatic skin lesions via neuroimmune interactions.
Authorship and Acknowledgments:
Ms. Chen Huang (senior experimentalist, School of Basic Medicine, Shanghai Jiao Tong University School of Medicine), Dr. Pei-Yi Sun (Department of Dermatology, Xinhua Hospital affiliated with Shanghai Jiao Tong University School of Medicine), and Dr. Yiming Jiang (Associate Chief Physician, Department of Otolaryngology, Renji Hospital affiliated with Shanghai Jiao Tong University School of Medicine) contributed equally as first authors. Prof. Tian-Le Xu and Dr. Xin Qi from Shanghai Jiao Tong University School of Medicine and Prof. Wei-Guang Li from our institute serve as co-corresponding authors.
This work was supported by various grants, including the Science and Technology Innovation 2030 Major Project (“Brain Science and Brain-Inspired Research”), the National Natural Science Foundation of China, and other Shanghai municipal-level funding. The study also benefited from collaborative support by Prof. Honglin Wang from Shanghai General Hospital, Prof. Zhirong Yao from Xinhua Hospital, Prof. Micheal X. Zhu from the University of Texas Health Science Center at Houston, and Prof. Yang Tian from East China Normal University.
Link to original paper: https://www.nature.com/articles/s41467-024-49577-3






 Address:No.138, medical college road
Address:No.138, medical college road  Postcode:200032
Postcode:200032  Tel/Fax:021-54237056
Tel/Fax:021-54237056 Emial:itbr@fudan.edu.cn
Emial:itbr@fudan.edu.cn